|
|
|
|
|
||
| SEARCH | SUBSCRIBE | ||
|
New Ideas
Energize Alzheimer's Battle
NY
Times, January 14, 2003
Dr.
Eliezer Masliah's finding that Alzheimer's was a disease of damaged
synapse connections has led to possible treatment strategies. Eventually,
there is the blank look, a sort of Alzheimer's stare, and a failure to
recognize a husband, a wife, a child. But the real question has been, What
is going on in the brain, and why do patients suffer the terrible
forgetfulness? For
years, the prevailing notion was that Alzheimer's was a disease of
brain-cell death. Pathologists could see that progressively, relentlessly,
brain cells died, leaving behind piles of debris that they detected in
autopsies and that are the hallmarks of the disease. But
now, many researchers are asking if that old hypothesis is correct. They
cite accumulating evidence that memory starts to fail long before brain
cells die, and that the disease, with its memory loss, begins as an
interruption of the signaling between living and healthy brain cells. If
they are right, it may be possible to stop Alzheimer's, and reverse the
memory loss, if treatments begin before brain cells die. The
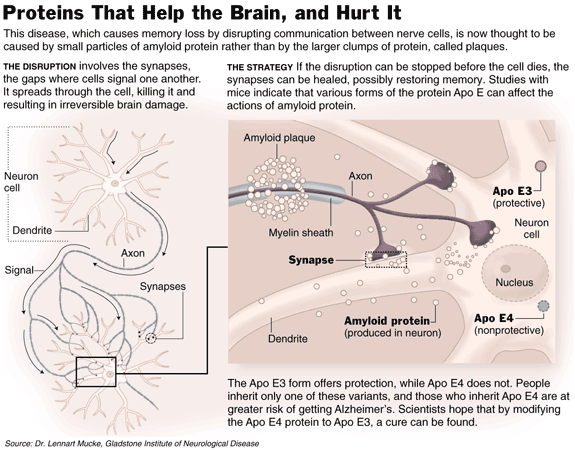
new idea is that the disease starts when small clumps of a protein,
amyloid, start interrupting signals between nerve cells in the brain. The
synapses, where one cell signals another in the brain's intricate
circuitry, no longer function. Yet,
at this early stage of the disease, researchers say, the nerve cells are
still alive and well, and the disease might be halted if the clumps of
amyloid could be removed or inactivated. But if the disease continues,
brain cells start to die. Dead and dying cells accumulate in heaps in the
brain, and the injured brain cannot be made whole.
The
work is still in a research stage, and the possible therapies are years
away, if they pan out at all. But Alzheimer's investigators say they are
invigorated because they have some tantalizing ideas of ways to stop the
small clusters of amyloid from inflicting injury, giving them hope that
they may be able to squelch the disease. "The
nice thing about synapses is that they are very plastic," said Dr.
Lennart Mucke of the Gladstone Institute of Neurological Disease and the
University of California at San Francisco. "As long as the cell body
is intact, nerve cells have a remarkable ability to rebuild their
synapses. It is much harder and often impossible to replace whole nerve
cells. But if we could interfere with synaptic damage early on, there is a
very good chance that cognitive function would come back."
Peter
DaSilva for The New York Times Dr.
Lennart Mucke and his colleagues at the Gladstone Institute are studying a
protein that may help protect the brain from dementia. In
a sense, researchers say, the evidence has been staring at them for years.
It began with a puzzle about the defining feature of Alzheimer's, amyloid
plaques, those microscopic patches of debris found in the brain. The
plaques, made up of large aggregates of amyloid protein, are unmistakable,
pathologists say. They look like tiny stars, about a tenth the thickness
of a hair and about five to six times as large as a brain cell. "One
of the main diagnostic features is the deposit of this insoluble material
in the brain," said Dr. Eliezer Masliah, a professor of neurosciences
and pathology at the University of California at San Diego. "This
gunk accumulates in the brain in very specific regions where memory and
functions like them happen, like in the frontal cortex and the hippocampus
where memories are recorded." For
years, he added, people thought this material was the main problem in
Alzheimer's. But,
in 1989, he and Dr. Robert Terry, who was chief of the laboratory there,
noticed a problem with the plaque hypothesis. They had brains from people
who had been severely demented and those with only mild memory impairment. They
expected to see many more plaques in those with severe dementia. They did
not. In fact, they found no tight link between plaque levels and the
degree of dementia. "We were quite surprised," Dr. Masliah said.
The idea that plaque caused dementia "had been such a dominating
force for so many years." Dr.
Masliah proposed another hypothesis. "I thought probably something
was going wrong at the level of the connections between the brain
cells," he said, reasoning that if the system was not working
properly, it might be because the communications between the nerve cells
were failing. Those connections, the synapses, which link the cells in
complex networks, might be damaged. But
his work was not widely accepted at the time. Why, colleagues asked, were
the synapses failing? Was there a link between synaptic damage and
dementia? Did it explain the lack of correlation between plaques and the
degree of dementia? In
the next decade, other scientists began finding small molecules that, they
proposed, might be the culprits in damaging nerve cells. The molecules
seemed to be clumps of amyloid protein, Dr. Mucke said, and a new picture
was emerging. These small amyloid clumps, he said, "do not just exist
in these garbage piles, the plaques, but they also float around like
cruise missiles." When they do, they might injure synapses. Different
investigators, stumbling upon various stages of the small clumps
independently, gave them different names. They say they struggled for
years to persuade colleagues to pay attention. Dr.
William L. Klein, a professor of neurobiology and physiology in the
Alzheimer's Disease Core Center at Northwestern University, called them
A.D.D.L.'s, for amyloid derived diffusible ligands, and reported that they
blocked communication between nerve cells. But, he said, when he first
presented his data at a meeting in 1998, the audience of scientists was
skeptical. "I overheard some folks saying, `How could that be? Why
didn't we see it?' " In
the meantime, Dr. Dennis J. Selkoe of Harvard was finding essentially the
same thing — chains of molecules, which he called protofibrils, that
disrupted nerve cell communication. The chains consisted of about 100
molecules of amyloid beta protein, compared with thousands in that make up
plaque in a fibril. Dr.
Selkoe added the small clumps of molecules to nerve cells in the
laboratory. "They killed them," he said, but the cells died more
slowly than they did when they were mixed with larger clumps that make
plaques. Then, Dr. Selkoe said, he injected protofibrils into rats and
found that they interfered with the first steps of memory and learning. In
the meantime, Dr. Mucke studied mice that he and his colleagues
genetically modified so that they produced human amyloid proteins. Long
before those proteins were deposited in plaques in the animals' brains, he
found, nerve synapses were damaged. As a consequence, nerve signals were
not being transmitted. Dr.
Masliah, his colleagues and Dr. John Morris, a neurologist at Washington
University in St. Louis, looked in the brains of people who had what might
have been the earliest stage of Alzheimer's, known as mild cognitive
impairment; those who had end stage dementia; and those of older people
who were not demented. When
Dr. Masliah examined the brains of people free of dementia, he found
healthy synapses. Brains of people with mild impairment had damaged
synapses, and the damage was even more pronounced in people with
Alzheimer's. That, he said, "suggests to me that Alzheimer's disease
is a disease of synaptic connections." It
also suggested a treatment strategy. "If
we believe that the actual plaques are the cause of Alzheimer's disease,
then we need to dissolve them and clear them from the brain," Dr.
Masliah said. "But
if we believe it is the precursor fragments, then we need to prevent those
fragments from being formed," he added. "The plaques are these
huge aggregates of smaller components. Imagine that we have a drug that
would break the plaque apart. Then you'd have all the little tiny
components floating all over the place and the situation would be worse.
So we should leave the plaques alone." Several
strategies might work, Dr. Mucke said. All involve interfering with
amyloid with the hope that treatment can be started, and the toxic protein
blocked, before it starts aggregating in large clusters, forming plaques,
and before brain cells die. One
possibility is to stop the production of amyloid with drugs, now under
development, that are akin to the protease inhibitors used to treat H.I.V.
Another
is to use the body's own immune system, stimulating the production of
antibodies that will attach themselves to the small amyloid clusters and
sweep them away. "You trigger the immune system into doing a cleanup
job," Dr. Mucke said. That is a strategy that works well in mice, but
initial studies in humans were halted because some of the patients
developed brain inflammations. "This
was not entirely unexpected," Dr. Mucke remarked. "Harnessing
the immune system is a tricky business and we have to learn how to
fine-tune it." It
also may be possible to enhance the body's own ways of removing the small
clusters of amyloid. There are enzymes, like one known as neprilysin, that
appear to chew up amyloid, Dr. Mucke said. "People make them
normally," he added, explaining that if their levels were increased,
it might tip the balance in people developing Alzheimer's, preventing them
from accumulating enough of the protein to damage their synapses. Yet
another idea is to try to help the brain cope with the small amyloid
proteins. The notion comes from studies linking a cholesterol-carrying
protein to a risk of developing Alzheimer's. People can inherit any of
three forms of the protein — Apo E2, Apo E3 or Apo E4. Those who have
the Apo E4 form are more likely to develop Alzheimer's than those with the
other forms of the protein. Dr. Mucke and his colleagues have found that
Apo E3 can protect the brains of mice from amyloid. (They have not yet
examined Apo E2, which confers an even lower risk than Apo E3.) "If
we look at middle-aged mice that make human amyloid and Apo E3, they are
fine," Dr. Mucke said. "At the same time, those that make human
amyloid and Apo E4 have lost a significant number of synapses." His
colleagues at the Gladstone Institute are developing drugs that convert
Apo E4 to apo E3. The two forms of the protein differ in just one amino
acid, No. 112 of the chain of 229 amino acids that make it up, and that
one amino acid determines how the protein is folded. They,
along with investigators at the University of California at San Francisco,
used computer modeling to screen large databases of chemicals for ones
that might fit into a groove in the Apo E4 protein and modify its shape so
it looks like Apo E3. They found several that looked promising. "We
are still at the cell culture level with these studies," Dr. Mucke
said. "But it would be one way of taking advantage of what nature
already has in place. We know that this protein has a big impact on
Alzheimer's disease. The idea is to take advantage of what nature is
revealing to us." But
would it work? Is there any hope that people can regain their memories
after the disease process has started? The
task ahead is not likely to be easy, Dr. Mucke and others caution. "We need to look at Alzheimer's disease as a disease that is as powerful and ferocious as cancer," Dr. Mucke said. "We need to fight it with equally heroic measures." But now, he said, "It is beginning to look like a tractable problem."
Copyright
© 2002 Global Action on Aging
|